生物体内的C-H键羟化反应,普遍通过含铁加氧酶来实现。在羟化反应中,加氧酶将氧分子转化为具有底物羟化能力的含氧活性中间体实现O2的活化。对于双铁羟化酶,目前经典的O2活化机制源于可溶性甲烷单加氧酶(sMMO),其特点是O2活化后形成菱形双四价Fe2IVO2中间体(Q),接着通过Q抓氢过程,羟化甲烷中惰性C(sp3)-H键(图1),该O2活化机制常被用于推测其它双铁羟化酶的反应机制。然而,最近实验研究发现,对于甲苯单加氧酶(T4MO)中苯环C(sp2)-H的羟化过程,其机制并非通过形成Q,而是在O-O键未断裂之前,O2活化形成的超氧物种进攻苯环(图1)。因此,探究C(sp3)-H键羟化过程是否还存在其它O2活化机制,是双铁加氧酶研究领域中一个重要的科学问题。
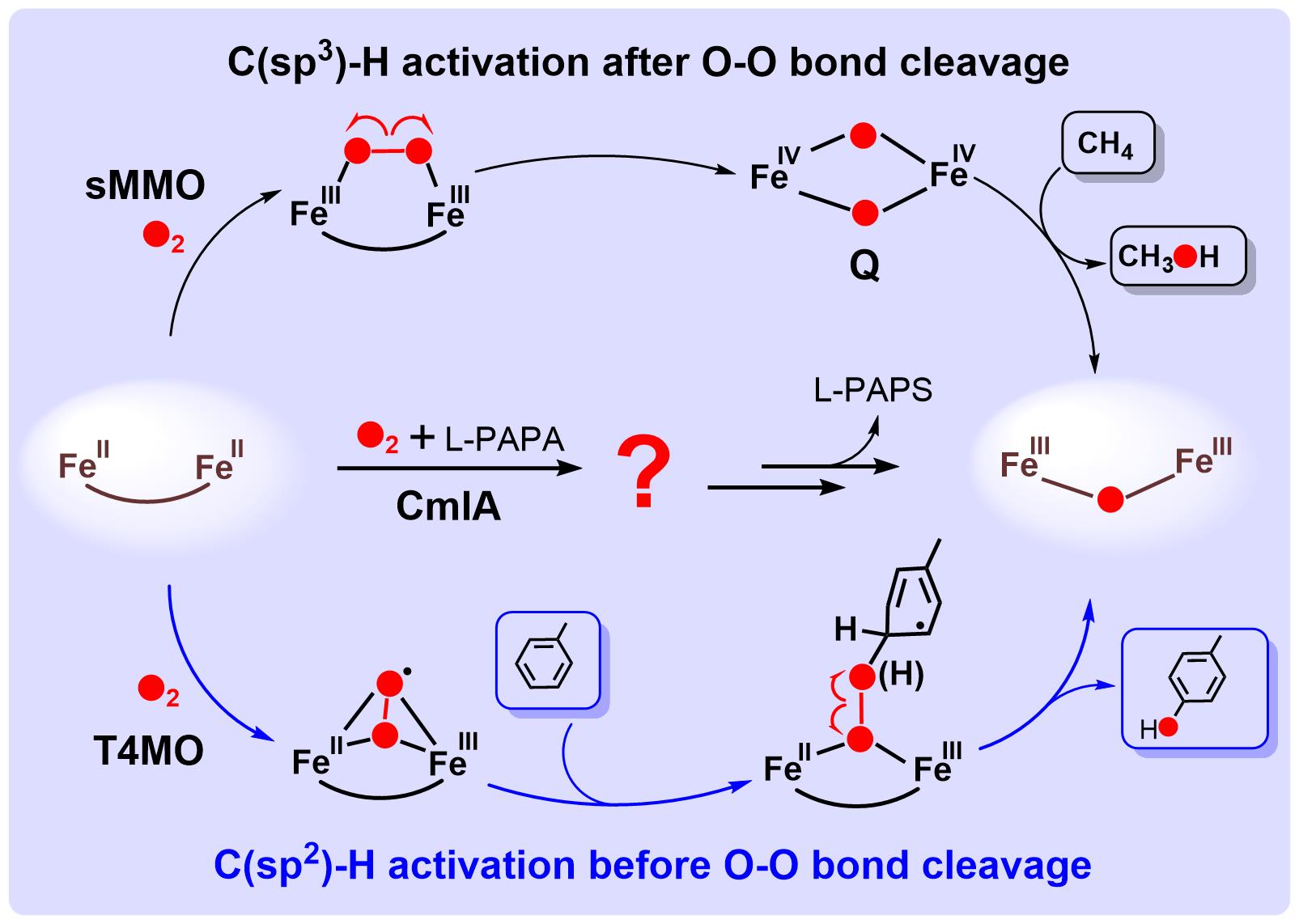
图1 双铁羟化酶的O2活化和C-H键羟化机制
在国家自然科学基金委、科技部、中科院的支持下,化学所光化学院重点实验室陈辉研究员团队在理论计算研究双铁加氧酶O2活化机理方面取得系列突破。在前期工作中(J. Am. Chem. Soc. 2017, 139, 13038-13046),他们利用自创的57Fe穆斯堡尔谱多尺度QM/MM模拟方法,成功确定了氯霉素生物合成中重要的双铁芳胺加氧酶CmlI的O2活化中间体结构(图2,左)。在后续黄素双铁蛋白Tm FDP活化还原NO的反应中(Angew. Chem. Int. Ed. 2019, 58, 3795-3799),他们用类似的多尺度模拟策略,首次揭示了黄素双铁蛋白中关键的双亚硝基中间体的第二配位层效应,及其对NO还原的重要影响(图2,右),从而将多尺度模拟方法的适用范围从确定O2活化过程第一配位层结构,成功拓展到探究NO活化过程第二配位层结构,为该类多尺度理论计算模拟方法在双核铁酶体系中更广泛的应用打下了基础。
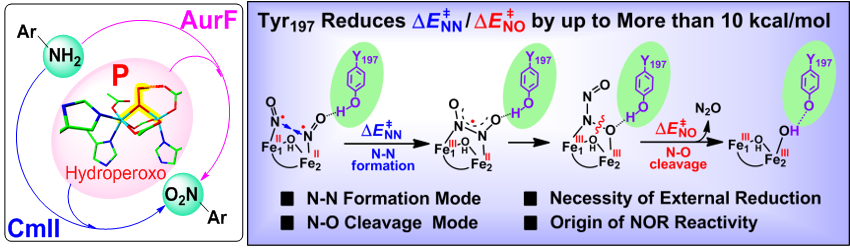
图2 通过理论计算预测得到双铁芳胺加氧酶CmlI中O2活化中间体结构(左),以及黄素双铁蛋白Tm FDP中NO活化中间体结构与反应机制(右)
最近,针对氯霉素生物合成中的第一步转化,即双铁羟化酶CmlA催化对氨基苯丙氨酸(L-PAPA)底物b位C(sp3)-H键羟基化反应,利用多尺度QM/MM模拟,陈辉研究员团队首次揭示了其既不同于sMMO,也不同于T4MO的O2活化新机制(图3)。研究发现,CmlA底物L-PAPA中待羟化的b位C(sp3)-H键邻位上自带的氨基,通过N-O成键,可以协助O2活化中O-O键发生异裂。这种氨基辅助的O2活化机制,产生了双三价Fe2IIIO中间体作为CmlA中真正的C(sp3)-H羟基化活性中间体,与双铁C(sp3)-H羟化酶sMMO中菱形双四价Fe2IVO2中间体Q存在本质的不同。由于CmlA中O2活化时O-O键必须先发生断裂,这在机制上也与双铁C(sp2)-H羟化酶T4MO完全不同。此外,研究还发现,O2活化过程中形成的N-O键,在C(sp3)-H羟化完成后,可以逆向发生断裂,从而使底物中的氨基得以再生,最终使氨基达到无痕协助O2活化的效果。最后,研究中发现的双铁第一配位层桥式羟基在O2配位后的重组现象,也为今后研究相关双铁加氧酶O2活化过程中发现新结构与机制提供了可能性。相关结果发表于Angew. Chem. Int. Ed. 2022, 61, e202211843。
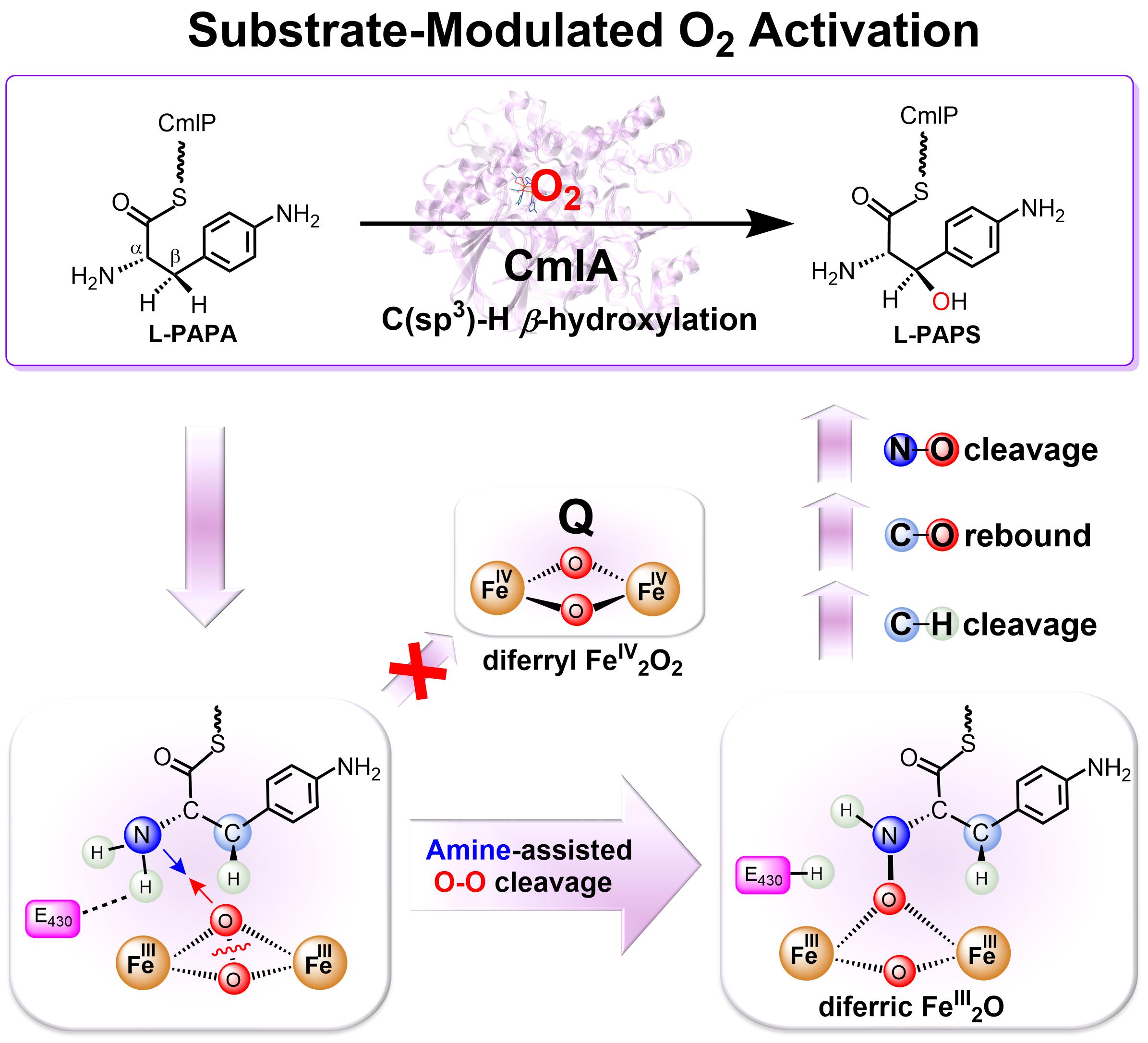
图3 理论计算揭示双铁羟化酶CmlA中独特的底物氨基辅助O2活化机制
光化学院重点实验室
2022年10月26日

